Post-Approval Compliance & Quality Assurance
Ensuring the ethical conduct of research goes beyond initial IRB review and continuing oversight—it requires ongoing attention to how studies are conducted in practice.
The Compliance Team conducts post-approval monitoring as part of its routine oversight of IRB-approved studies. These reviews aim to improve research practices and protect research participants’ rights and welfare through IRB Office guidance, resources, and educational support. However, the entire Northwestern HRPP, research teams included, is responsible for the conduct of compliant human research, which can be facilitated by internal Quality Assurance (QA) reviews.
This page provides researchers with essential guidance to support them in conducting QA reviews, particularly through the use of the IRB Office's post-approval monitoring (PAM) checklists as a structured compliance tool.Using the Post-Approval Monitoring Checklists during an internal Quality Assurance Review
The Compliance Team makes the same post-approval monitoring checklists used during the IRB Office’s monitoring available to researchers as a resource. The Compliance Team recommends that researchers use the post-approval monitoring checklists outside of routine IRB reviews to help ensure their study complies with applicable federal regulations, state laws, institutional policies, and best practices.
When incorporated into internal QA processes, the checklists provide a structured and effective framework for ongoing oversight throughout the life of the study, while also offering several additional benefits for the research team:
- The checklists may serve as documentation of ongoing oversight of study conduct.
- The checklists can help prepare study teams for quality assurance visits by external monitors or auditors.
- The checklists reflect what the Northwestern University IRB Compliance Team would expect to see when performing a for-cause (directed review) or routine review of the research study.
When to Conduct a QA review
- At study startup: Conduct a routine QA review using the checklists after a) the Principal Investigator has obtained IRB approval, b) the study team has completed required training, c) study files are organized, and d) the first participant has been enrolled. This review helps confirm that regulatory and study procedures are in place and compliant at the beginning of the study, preventing larger issues down the road.
- Mid-point of study: Conduct routine QA reviews quarterly or bi-annually, or as appropriate based on the pace of enrollment, risk level, and complexity of the study. These periodic reviews help maintain ongoing compliance and may identify areas requiring corrective action.
- End of Study: Conduct a routine QA review to ensure that outstanding issues (e.g., unresolved deviations, missing reports, or incomplete study documentation) are addressed, and the study is ready for IRB closure and record retention in accordance with institutional and regulatory requirements.
- For-Cause: Conduct a for-cause QA review in response to unanticipated events (e.g., serious, suspected, or repeated non-compliance, safety risks to participants, or data integrity issues) or to prepare for a scheduled external audit.
What to do After Completing a QA review
- Report: If the review uncovers a finding, assess whether the information meets the IRB's reporting criteria and, if so, submit the information, as appropriate, to the IRB. Review the Reportable New Information (RNI) webpage.
- Document: Create and retain documentation of the QA review, including all findings, assessments, and any corrective or preventive actions implemented as part of post-approval monitoring. Non-compliance or deviations that do not rise to the level of reporting to the IRB should still be documented.
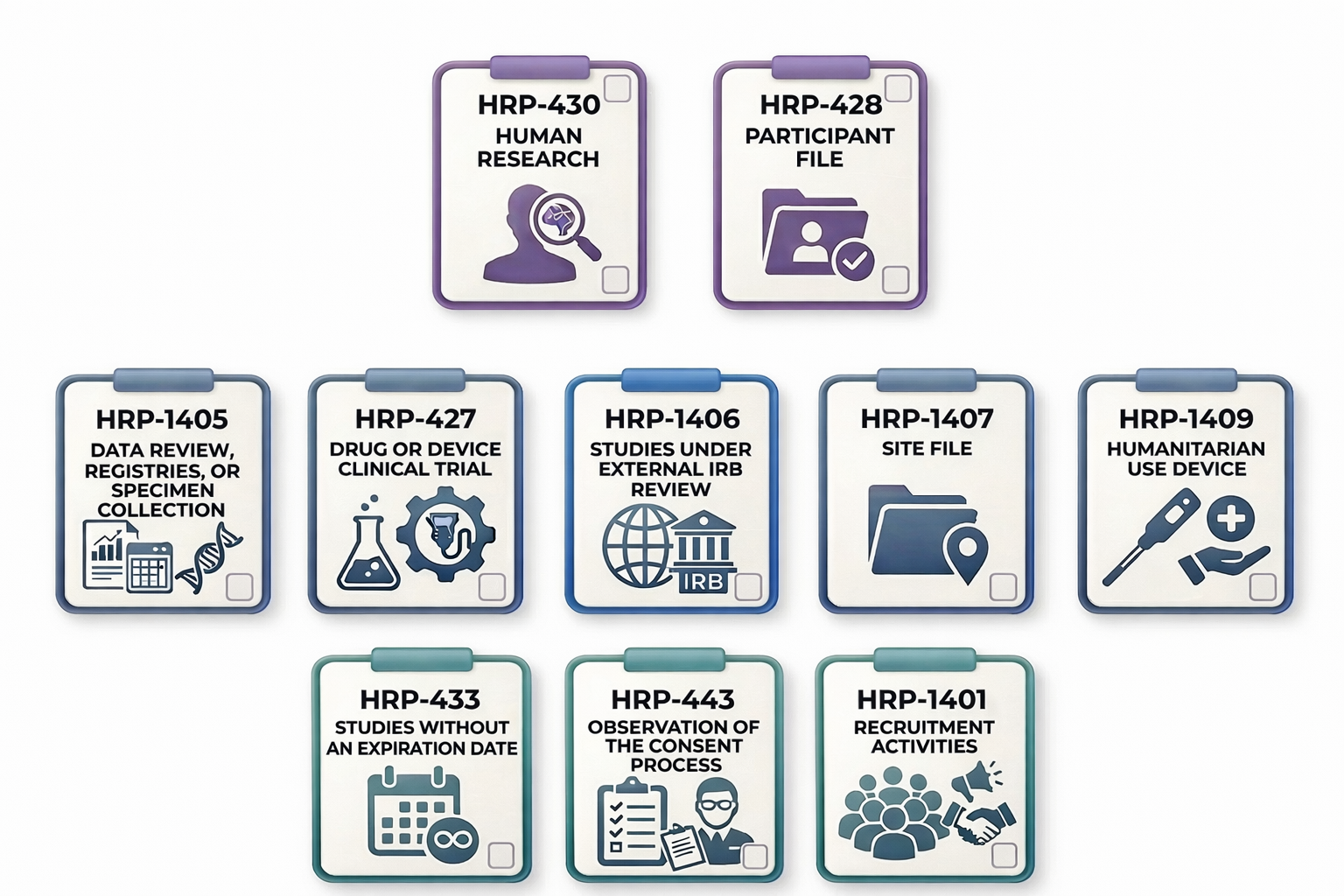
Types of Post-Approval Monitoring Checklists
The extent and nature of the QA review should be planned for each study, taking into account factors such as study complexity, risk level, enrollment goals, etc. Based on these considerations, the study team should select the appropriate post-approval monitoring checklist(s) outlined below, noting that more than one checklist may be applicable and beneficial depending on the study’s design and risk profile.
The Compliance Team's post-approving monitoring checklists are available for download and use on the Checklists & Worksheets webpage.
Foundational Checklists
The two post-approval monitoring checklists listed below are considered foundational for non-exempt IRB-approved research studies because they address core regulatory requirements, study oversight responsibilities, and participant protection expectations that apply across most study types and phases.
Human Research (HRP-430)
- For use in non-exempt studies.
- Use this checklist in combination with applicable supplemental PAM checklists listed below, as appropriate based on the status and scope of the research.
Participant File (HRP-428)
- For use when the Principal Investigator has initiated a medical record review, specimen collection, and/or has consented and enrolled participants.
Supplemental Checklists
The following post-approval monitoring checklists are designed to supplement the foundational checklists to address additional considerations for specific types of research activities. Researchers should use the checklist(s) that best align with their study’s design, procedures, and oversight requirements in addition to using the foundational checklists.
Data Review, Registries, or Specimen Collection (HRP-1405)
- For use if the research involves medical record (data) reviews, registries, and/or specimen collection and retention.
Drug or Device Clinical Trial (HRP-427)
- For use if the study is a clinical trial and involves an investigational drug or device.
Studies Under External IRB Review (HRP-1406)
- For use if the study relies on an External IRB for review.
Site File (HRP-1407)
- For use if external study sites, excluding Northwestern affiliates, rely on the Northwestern IRB for review.
- Complete one Site File checklist for each site that relies on Northwestern IRB.
Humanitarian Use Device (HRP-1409)
- For use if the study involves treatment plans with a humanitarian use device (HUD).
Stand Alone Checklists
Each of the following three post-approval monitoring checklists is a stand alone QA tool designed to evaluate a specific research activity. Each of these checklists functions independently to represent other types of research procedures separate from the above types of checklists.
Studies Without an Expiration Date (HRP-433)
- For use when conducting a focused Study Status Assessment on non-exempt, minimal risk studies approved or renewed by the IRB with no expiration date.
- Researchers may use this checklist to complete an internal review of the study status in place of submitting an annual Continuing Review to the IRB.
- It is recommended that the Principal Investigator (PI) or a designated team member conduct this internal review annually or every 2-3 years while the study is open.
Observation of the Consent Process (HRP-443)
- For use when conducting a QA review of the informed consent process in real time, to ensure the process uses clear communication and respects participants’ rights to informed decision-making.
- If used, the participant must verbally agree ahead of time to allow the observation of the consent process by the observer. If the participant does not agree, the consent process may proceed without an observer.
Recruitment Activities (HRP-1401)
- For use if the study utilizes recruitment advertisements, to assess whether the recruitment procedures follow regulatory and institutional requirements.
Recommendations to Address Common Observations
Understanding common observations can help facilitate effective QA reviews. Below are examples of common observations that may arise, and best practice recommendations on how to address them. In addition, assess whether any observations meet the criteria for IRB reporting.
Observation: The consent form contains the required signatures, but there is insufficient documentation of the process to obtain informed consent
Recommendation: Implement a consent process checklist to use in real time while obtaining consent that ensures the consent process for each participant was conducted per
SOP Informed Consent Process for Research (HRP-090) and is documented in the participants’ records.
Observation: Identification of protocol deviations, missing study documentation, or other study issues
Recommendation: Recover and file missing documentation where possible, document past deviations and unrecoverable documentation in Notes-to-File, and record deviations in real time on a protocol deviation log moving forward. Use the IRB Office’s
Study Support Resources and Templates as needed.
Observation: Enrollment is complete and identifiable data is no longer accessed or analyzed, but the study is still open with the IRB
Recommendation: Assess if remaining activities meet the definition of human research, review IRB study closure requirements, and confirm that all required regulatory and study documentation is complete and up to date to support IRB closure. Use the Studies Without an Expiration Date Checklist (HRP-433) as a guide. If appropriate, create a timely submission in eIRB+ to request IRB approval of study closure.
Observation: The mechanism to document participant/chart/specimen eligibility is unclear
Recommendation: Implement an eligibility checklist or tracking log to document criteria assessed during screening and enrollment for each participant/chart/specimen, and confirm the documentation is stored in the PI’s study record, to demonstrate protocol adherence and data integrity.
Observation: There are out-of-date funding source(s) and study personnel in the eIRB+ study record
Recommendation: Confirm that all study personnel have completed and maintained required HSR training during their study involvement. Confirm that all documentation (i.e. CITI completion reports or certificates) is retained in the PI’s study record, and that up-to-date HSR training dates are reflected in eIRB+.
Observation: Study personnel have expired Human Subjects Research (HSR) training.
Recommendation: Confirm that all study personnel have completed and maintained required HSR training during their study involvement. Confirm that all documentation (i.e. CITI completion reports or certificates) is retained in the PI’s study record, and that up-to-date HSR training dates are reflected in eIRB+.
Contact Us
The Compliance Team works closely with researchers to navigate federal regulations, state laws, and institutional policies, ensuring studies meet all requirements to protect human participants. This partnership approach reflects our belief that safeguarding research participants is a shared responsibility across the HRPP, the IRB Office, and our research community.
Our team is here to help - whether you have a quick question or need more in-depth guidance. We are always available for a one-on-one Zoom or phone consultation, or via email.
Contact the IRB Office Compliance Team at irbcompliance@northwestern.edu or call 312-503-1376 with any questions.